The entire human genome is made up of 6.4 billion nucleotides, which are individual positions in the human DNA denoted by the letters ‘A’,’T’,’C’ and ‘G’. In comparison, the SARS-CoV-2 genome is much smaller, with only about 29,000 nucleotides. This week, QBRI experts discuss how variations in the human host and viral genome sequence may determine the risk and severity of COVID-19.
Mutation of SARS-CoV-2
Mutations, the changes in the genetic sequence, are a natural part of a virus life cycle. A mutation occurs randomly, inherits to all copies of the next generation, and rarely impacts an outbreak (1). While some mutations are ‘silent’ and may not impact the viral biology, others may directly affect the structure of the virus and, hence, have biological implications on the viral infection or replication rates.
SARS-CoV-2, the RNA virus responsible for the COVID-19 pandemic, also evolves by mutation. Knowledge of the mutation rate in SARS-CoV-2 would help us answer key questions such as: How fast do mutations occur? or Where precisely in the genome do mutations occur?
This information is critical in designing an effective drug for treatment. There have been global initiatives to share the viral genome that will aid scientists across the world to infer the evolutionary history of SARS-CoV-2 in real time.
The Global Initiative on Sharing All Influenza Data (GISAID) and Nextstrain are such initiatives spearheading the evolutionary tracking analysis of COVID-19. These tracking systems would give extensive information about the different outbreaks occurring in a community, eventually helping public health officials to contain the viral spread.
To understand and trace the evolution of SARS-CoV-2, it is important to estimate its mutation rate. In general, the estimated mutation rates in coronaviruses are moderate to high compared to other single stranded RNA viruses (2). Based on current data, it seems that SARS-CoV-2 mutates much more slowly than the seasonal flu. SARS-CoV-2 has a mutation rate of less than 25 mutations per year compared to 50 mutations per year for seasonal flu.
The entire genome or genetic content of SARS-CoV-2 consists of 29,903 nucleotides - the individual positions or building blocks that constitute the genome. A recent report shows the prevalence of single nucleotide mutation transition as the major mutational type across the world and identified at least three clades or evolutionary groups characterized by geographical and genomic specificity (3). These clades were named as “G” (Europe, Africa, Americas and Oceania), “V” (Europe) or “S” (Asia and Americas) groups based on the mutations D614G (spike protein), G251V (ORF3a) or L84S (ORF8) observed in the SARS-CoV-2 sequence.
Another research study identified six major subgroups of SARS-CoV-2 strains with strong geographic preferences. These groups were classified as 1) Europe-1 with a majority from Iceland; 2) Oceania/Asia including Australia and China; 3) Americas with a majority from the United States; 4) Europe-2 including Great Britain and Iceland; 5) Asia-1 with a majority from China; and 6) Asia-2 including China and Japan (4).
Another recent study of SARS-CoV-2 indicated that some genetic changes can influence the virulence and transmissibility of SARS-CoV-2. This study revealed that four combined mutations can probably increase virus transmissibility. These variants were prevalent in Europe, where apparently COVID-19 infection was more severe than other regions, and are located in genome locations coding for proteins necessary for viral infection and vaccine efficacy (5) (Figure 1).
 or genetic variations")
Figure 1. Single nucleotide polymorphisms (SNP) or genetic variations
The high mutation rate aids the seasonal flu virus to evade vaccines, while the significantly slower mutation rate of SARS-CoV-2 gives us hope for the development of potentially long-lasting vaccines against the virus and effective treatment of patients across different geographical locations.
In a recent study, mutation burden has been positively correlated with the fatality of COVID-19 patients (4). Moreover, as the existing mutations continue to accumulate in time and expand globally (Figure 2), new mutations will constantly emerge to enrich the diversity of mutations (6, 7). Therefore, continuous isolation and sequencing of the viral genome is an important criterion in the battle to contain the pandemic and strategize the development of vaccines and anti-viral treatments (8).
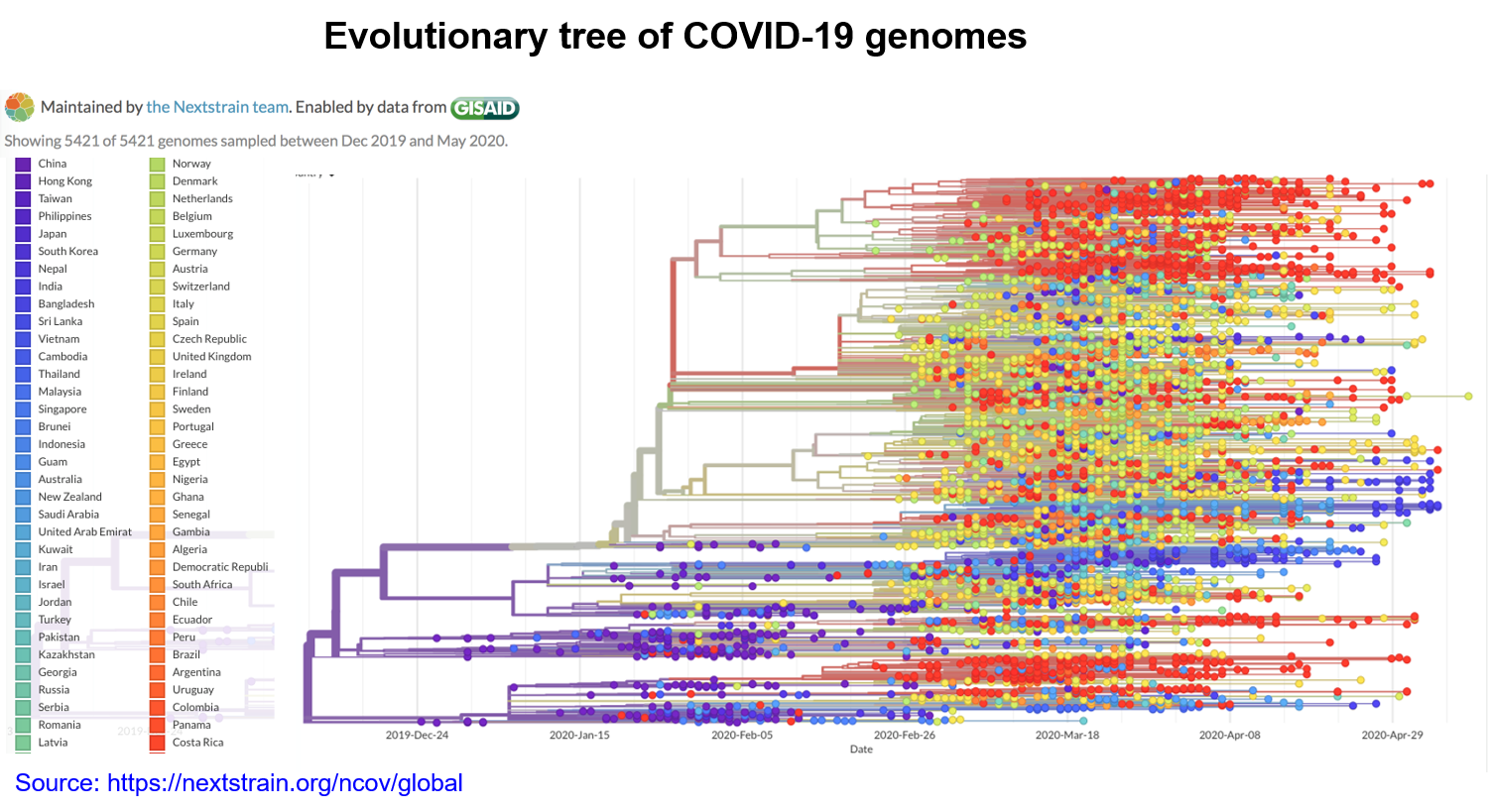
Figure 2. The SARS-CoV-2 phylogenetic tree. The left-most branch in the tree represents a mutation common to all sequences corresponding to the older strain of the virus. Colored circles denote mutations the virus has accumulated over time. Branches are added to the right to depict new mutations. A vertical line indicates identical sequences. The colors denote regional branches of the tree.
Genotyping as an Accessory COVID-19 Diagnostic Tool
Genotyping is the technology that detects small variations in the genetic sequence, known as single nucleotide polymorphisms (SNPs) that can lead to both physical differences, that make us unique, and pathological changes that lead to diseases (figure 3). Human genome genotyping has given rise to the fields of pharmacogenetics and personalized medicine, which focus on designing individualized therapies and specific synthetic drugs based on the genotype of each individual. It also allows the study of individual predisposition to develop certain diseases, the disease severity or their response to treatments.

Figure 3. Genotyping can identify our genetic uniqueness
COVID-19 symptoms range from very mild to severe. To better understand the inter-individual variability in the response to SARS-CoV-2 infection, genotyping may prove useful. Interestingly, polymorphisms or variations in the Human Leukocyte Antigen (HLA), a protein on the surface of human cells that the immune defense system uses to recognize ‘self’ or ‘foreign’, have been shown to play a role in the spread and severity of COVID-19 (9).
In addition, blood group variants have been associated with COVID-19. A first pre-print study was published by researchers in China who studied a particular variant (SNP rs505922) in the ABO gene that determines the blood group, and COVID-19 susceptibility (10).
A second report from a genome-wide association study (GWAS), conducted by different Italian and Spanish centers, found an association between blood group type and severity of COVID-19. Their meta-analysis of around 8.5 million SNPs revealed that some variants may help to identify severe COVID-19 cases. Importantly, according to this study, people with blood group O have a protective effect against COVID-19 (11). More recently, 23andMe, a company that offers a direct-to-consumer genetic testing service, reported unpublished preliminary results enrolling around 750,000 subjects also suggesting that the O blood type can be protective against the virus.
On the other hand, genotyping is also an effective method of identifying variants and mutations in the genome of viruses, as described earlier to control the spread of the pandemic and to locate the origin of the foci of infection. This technology is important in clinical research for the characterization of genes associated with diseases in an area of science often referred to as molecular epidemiology or forensic microbiology.
Genotyping of SNPs are accurate and sensitive. However, some current technological limitations are present, as almost all genotyping tests are partial, time-consuming and involve a multi-step process. Genome-wide association studies and mass-sequencing technologies, like DNA sequencing and microarrays, may overcome some limitations, providing the whole-genome genotyping and identification of novel variants.
Section Contributors
Mutation of SARS-CoV-2: Dr. Nishant N. Vaikath (Research Associate, QBRI)
Genotyping as an Accessory COVID-19 Diagnostic Tool: Dr. Salam Salloum Asfar (Postdoctoral Researcher, QBRI)
Arabic text validation: Dr. Nour K. Majbour (Senior Research Associate, QBRI)
Editors: Dr. Adviti Naik (Postdoctoral Researcher, QBRI) and Dr. Alexandra E. Butler (Principal Investigator, QBRI)
For references, please click here.
Share:
Related News